- Viewed - 2128
- Printed - 0
- Emailed - 0
- PDF Downloaded - 1013
A Clinopathological Correlation of Medullary Thyroid Carcinoma
Year : 2020 | Volume : 48 | Issue : 0 | Page :
MJWI.2022/94
Kalpana Ketan Kulkarni , Savita Sanjay Patil , Monika Singh ,
Date of Web Publication 31-Mar-2022
Keywords
Medullary Thyroid carcinoma, Multiple endocrine neoplasia 2B, Amyloid
ABSTRACT
Keywords: Medullary thyroid carcinoma, Multiple Endocrine Neoplasia 2B, Amyloid
Introduction:
Medullary thyroid carcinoma (MTC) is a rare thyroid malignancy associated with a higher incidence of distant metastasis and poorer prognosis compared with the more frequently encountered well-differentiated papillary and follicular thyroid carcinomas. In addition, 25% of MTCs occur as hereditary forms, the majority of which occur as part of the multiple endocrine neoplasia (MEN) 2 syndromes.1
MTC originates from the parafollicular C cells unlike other the more common thyroid malignancies that arise from follicular cells.1,2 MTC occurs as a sporadic tumor in about 75%–80% of those treated and as a hereditary tumor in the rest.3
There are two types of hereditary MTC. One is isolated familial medullary thyroid carcinoma (FMTC) and the other is as a part of Multiple Endocrine Neoplasia 2(MEN2). MEN2 is further subdivided into type 2A and type 2B.4 The Multiple Endocrine Neoplasia 2B(mucosal neuroma syndrome, Wagenmann-Froboese syndrome)4is an autosomal dominant syndrome characterized by medullary thyroid carcinoma, pheochromocytoma, mucosal neuromas, ganglioneuromatosis of the gut and marfanoid habitus.5,6 MEN2B is a rare syndrome, and its phenotype may not always be noted by the physician.
MTC is microscopically composed of nests and sheets of polygonal or spindloid cells separated by fibrous septae. Amyloid deposits within the stroma are characterstically found.7
In this article, we illustrate the clinicopathological correlation of sporadic and hereditary MTC. We also emphasize the importance of recognizing hereditary MTC.
[
[
[
Materials and methods:
This is a retrospective, cross sectional, descriptive study from a single tertiary care center. This study was approved by Ethical Committee of the institute. This study spans duration of 6 years from January 2013 to December 2019. Clinical data, tissue blocks and hematoxylin and eosin stained slides of MTC was retrospectively retrieved from the archives of pathology department. A total of 6 cases of MTC were identified from records. The data was evaluated and studied for clinicopathological correlation. The slides were reviewed by two pathologists independently. Immunohistochemical stain for calcitonin was done in one case.
Results:
During the study period, a total of 37963 surgical specimens were received. Of these, 319 were thyroid specimens. Non neoplastic thyroid comprised 232 of 319 cases (72.7%) which included colloid goiter, multinodular goiter, Hashimoto's thyroiditis, colloid cyst and others. Neoplastic thyroid resections comprised 87 out of 319 cases (27.3%) of which 36 were benign thyroid follicular adenoma (41.3%) and 51 were malignancies (58.7%). Of the total malignancies, papillary and follicular thyroid carcinoma was common. 23 cases of papillary carcinoma (45.1%), 14 were follicular carcinoma (27.5%), 7 were of MTC (13.7%) and 7 cases were of other rarer thyroid malignancies and thyroid metastasis (13.7%). Of the 7 cases of MTC, 6 were sporadic (85.7%) and 1 was hereditary (14.3%).
Among the sporadic cases of MTC, four cases were male (66.7%) and 2 were female (33.3%).The mean age at diagnosis was 41.5 years (range 28- 73 years). All presented with neck swelling. In the preoperative evaluation, all of the patients underwent fine needle aspiration cytology (FNAC) with diagnosis of positive for malignancy was rendered in 5 of 6 cases (83.3%) with exact subtyping of MTC given in 3 cases (60.0%). Repeated FNAC in one case was inconclusive (16.7%). Total thyroidectomy was performed in 5 cases (83.3%). One case (16.7%) where the FNAC was inconclusive, hemithyroidectomy was done followed by total thyroidectomy. Grossly, tumor was unifocalall the cases. The mean tumor diameter was 2.6 cm (maximum being 4 cm and minimum being 2 cm). Tumor was located in right lobe in 4 cases (66.7%), in left lobe in 2 cases (33.3%).The tumor was relatively well circumscribed in all the cases and the on cut section showed tan white color. Areas of hemorrhage and necrosis were noted in 2 cases (33.3%).In our study, patients were diagnosed on the basis of histopathology, which is the gold standard for diagnosis.Microscopically, the most common architectural pattern noted was nests and sheets in all of the cases. Additional patterns noted were follicular pattern in one case (16.7%) and trabeculae in one case (16.7%). Individual cells were round to oval in all the cases, plasmacytoid in 3 cases (50.0%) and spindloid in 2 cases (33.3%). Mitotic figures were infrequent. Amyloid was present in all cases (100%). Lymph node resection was done in 4 cases (66.7%) with lymph node metastasis noted in all of them. No lymph nodes were received in 2cases (33.3%).
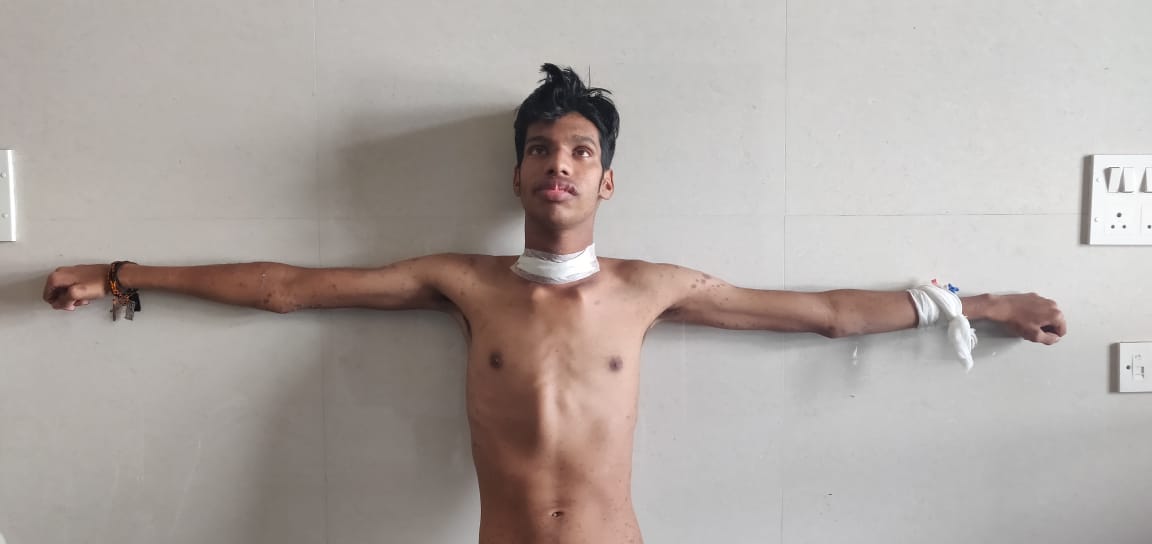
The hereditary case was a 23 year old young male patient. He presented with skin rashes and was found to have marfanoid features (tall and slender stature, an arm span length more than body height, high arched palate, elongated face, adducted thumb projecting beyond ulnar border in clenched hand), thickened upper eyelids, swollen lips along and thyroid swelling. Total thyroidectomy specimen was received. Grossly, the tumor was characteristically multifocal and bilateral, largest nodule measuring 3 cm in diameter. On cut section, the tumor was greyish white in color. Lymph nodes were not received. Microscopically, tumor was arranged in nests and sheets with presence of amyloid. Immunohistochemical staining with calcitonin was done and was positive.
Although the family history was negative for any thyroid malignancy, MEN2B4, 5 was diagnosed based on the presence of characteristic phenotype (Marfanoid habitus, thickened upper eyelids, swollen lips) with multifocal and bilateral MTC in a young male. A further evaluation for presence of pheochromocytoma was done and found to be negative. Follow up was advised.
Discussion:
MTC, a rare thyroid malignancy, that represents less than 10% of all thyroid cancers8-10, has a different clinicopathologic behavior than others such as papillary and follicular cancers. It secretes calcitonin and may occur either as a hereditary or a sporadic entity. In our study, we found MTC in 13.7% of all thyroid malignancies.
The mean age at diagnosis was 41.5 years, which is a decade lower to what has been reported elsewhere.11In other studies MTC was found to be more common in women.12 In our study, MTC was more common in men (66.7%). These discrepancies from other studies can be attributed to our small sample size.
MTC occurs about 75%–80% of the time as a sporadic tumor and 20%–25% as a hereditary tumor.2, 3 In our study, out of the total 7 patients, 6 patients (85.7%) had the sporadic form of the disease and 1 patient (14.3%) had a hereditary type.
Biochemical screening includes measurement of calcitonin levels and genetic screening includes testing for RET mutation studies.13 However, in our study preoperative calcitonin levels and RET mutation studies were not done.
Accurate cytological diagnosis of MTC by FNAC was given in 55-61% of cases in different studies.14It is known that MTC has a variable appearance on FNAC, which often results in a doubtful diagnosis. In our study, malignancy was identified in 83.3% of cases with exact subtyping of MTC given in 60.0% by FNAC.
MTCs are usually unifocal, solid well-circumscribed tumors of variable size15 which was consistent with our study. Microscopically, the MTC is characterized by a nests and sheets of tumor cells. Cells are mainly round, polygonal or spindloid with few studies reporting other patterns like follicular, trabecular, oncocytoid, etc.16-19 This was also the most common pattern in our study. Other patterns noted in our study were follicular pattern in one case (16.7%) and trabeculae in one case (16.7%). Necrosis and hemorrhage appear more frequently in larger tumors.16-19 In our study, necrosis and hemorrhage noted in 2 cases (33.3%). Stromal amyloid deposits may be seen in up to 80%. Presence of amyloid is a unique feature which is not seen in other thyroid tumors.16, 19 In our study, amyloid was present in all the cases (100%).
The hereditary MTC is rare and can be seen as familial medullary thyroid carcinoma(FMTC) or as a component of MEN2A and MEN2B. MEN2B is inherited autosomal dominant syndrome that includes mucosal neuromas(100%), ganglioneuromastosis(100%) and Marfanoid habitus(common), MTC(90%) and pheochromocytoma (45-50%).20-21 The clinical diagnosis of MEN2B could be done by identification of characteristic phenotype with peculiar facial features like thickened lips, elongated face, a ‘Marfanoid’ body habitus with tall stature and long limbs, mucosal neuromas(of lips, oral mucosa, tongue, eyelids, palate and intestinal mucosa) in addition to MTC and Pheochromocytoma.
In our study, the hereditary MTC case presented with facial features like thickened upper eyelids, swollen lips, Marfanoid habitus (tall stature and hyperflexible small joints) and MTC. With the characteristic phenotype and a histopathologically confirmed MTC, a diagnosis of MEN2B was given. Hereditary MTC occurs at a younger age group, with our case presenting at an age of 23 years.11The Hereditary MTCs are often bilateral and multifocal.22-23In our study, the single hereditary case presented as bilateral and multifocal thyroid tumor. Microscopically, similar morphology is seen in both sporadic and hereditary forms.
As there can be a coexisting pheochromocytoma in these patients of MEN2B, it is important to carefully search for pheochromocytoma before operating for MTC. Missing a diagnosis of MEN2B in such cases can lead to catastrophic intraoperative complications. Hence, we recommend evaluation of all young MTC patients for presence or absence of pheochromocytoma. In our case, though no suspicion of MEN2B was kept before operation, fortunately there was no pheochromocytoma. Regular follow up of the patient did not reveal any other malignancy.24, 25
In our study, there are certain limitations. We had a small sample size. This study being retrospective, all clinical details were not available. Also, RET mutation and preoperative calcitonin levels were not available.
Conclusion
MTC is a rare thyroid malignancy (13.7%) with unique clinicopathological features compared with other thyroid malignancies. FNAC can suggest a diagnosis of MTC (60.0%) even before histopathology. This helps in preoperative evaluation of patients.
MTC associated with MEN2B is rarer and seen younger age with a characteristic phenotype. As the syndrome is rare, the phenotype may be missed. Even those patients with MEN2B who present with the typical phenotypic features may not be recognized and followed up as a sporadic case. And hence, we recommend evaluation of patients with MTC, especially at a young age, for hereditary syndromes. We also recommend regular follow up of the case and proper screening of family members.
References